Data di ultimo aggiornamento 14 marzo 2024
Italiano | English
The in vitro diagnostic medical device sector is going through a transitional period in which some provisions set out by the Regulation (EU) 2017/746 (IVDR) such as the registration of economic operators and in vitro diagnostic medical devices, although mandatory from the date of application of the Regulation, cannot be fulfilled according to the method provided by the IVDR until the European Database Eudamed is not fully functional (as provided for in Article 113, para. 3, letter f).
In this context, in order to meet registration obligations in Italy it is required the registration in the Repertorio dei Dispositivi Medici (RDM).
Manufacturers and authorised representatives established in Italy which place in vitro diagnostic medical devices on the market under their own name are obliged to notify to the Ministry of Health their data and those identifying their devices, in accordance with the provisions of Article 10, paragraphs 1 and 3 of Legislative Decree 332/2000.
As reported in Art. 30 paragraph 3 of Legislative Decree 138/2022, article 10 of Legislative Decree 332/2000 is repealed twenty-four months after the date of publication of the notice that Eudamed is fully functional.
In the same way for all IVDR-marked in vitro diagnostic medical devices manufacturers and authorised representatives established in Italy which place in vitro diagnostic medical devices on the market under their own name are obliged to report to the Ministry of Health their data and those identifying their devices.
Art. 10, paragraph 2, also requires that for devices listed in Annex II of Legislative Decree 332/2000 and devices for self-testing, placed on the market in Italy (involving a Notified Body for certification purposes), the manufacturers and/or authorised representatives, also not established in Italy, should send to the Ministry of Health, in addition to the communication of identification data, also the labels and instructions for use.
In the same way for all IVDR-marked in vitro diagnostic medical devices that require the issuance of a certificate by a Notified Body (Class B, Class C and Class D), placed on the market in Italy, it is required that manufacturers, even if not established in Italy, should send the Ministry of Health, in addition to the communication of identification data, also the labels and instructions for use.
The manual of the instructions for use and the label may be translated into one or more official languages of the European Union among which the Italian language version must be included as set in Annex I, section B, point 8 of Legislative Decree 332/2000 and art. 6 paragraph 2 of Legislative Decree 138/2022.
As reported in Art. 30 paragraph 3 of Legislative Decree 138/2022, article 10 of Legislative Decree 332/2000 is repealed twenty-four months after the date of publication of the notice that Eudamed is fully functional.
From 5 June 2014, with the Decree 23 December 2013, manufacturers and authorised representatives of in vitro diagnostic medical devices, or their delegates, fulfil their obligations of notify to the Ministry of Health by registering in the Repertorio dei Dispositivi Medici.
Similarly, for devices intended for performance evaluation, manufacturers and authorised representatives based in Italy, or their delegates, shall comply with the provisions of Legislative Decree 332/2000 by registering in the Repertorio dei Dispositivi Medici using the functionality available in the Dispositivi Medici area under the menu item “Dispositivi in valutazione delle prestazioni PE”.
For manufacturers or authorised representatives which registered their devices before 5 June 2014, using the methods in use before the entry into force of the Ministerial Decree of 23 December 2013, there is no obligation to repeat registrations according to the current method (registration in the RDM). Data recorded before 5 June 2014 were not migrated to the Repertorio dei Dispositivi Medici due to the significant differences between its data structure and the pre-existing IVD database.
Manufacturers and authorised representatives who need to make changes to information registered before 5 June 2014, as they can no longer change the data, must proceed with the registration in the Repertorio dei Dispositivi Medici. Changes include the case of devices that are marked in accordance with the IVDR.
Each device marked pursuant to the IVDR must have a different number of Repertorio from the same device marked pursuant to Legislative Decree 332/2000.
Registration in the Repertorio can be done voluntarily even if the registration obligations, prior to 5 June 2014, under Article 10 of Legislative Decree 332/2000 were fulfilled, for the purpose of participating in supply tenders promoted by National Health Service (NHS) facilities and the subsequent establishment of business relations.
According to the Decree 23 December 2013, facilities directly managed by the National Health Service shall refrain from requesting from suppliers any information that is declared by them to be available in the Repertorio dei Dispositivi Medici and up-to-date on the date of the declaration.
Registration of an in vitro diagnostic medical device in the Medical Devices Database/ Repertorio dei Dispositivi Medici can be requested by:
The person who directly carries out transactions in the Medical Devices Database/ Repertorio dei Dispositivi Medici is configured as Declarant and is always a natural person (Italian or not). Only declarants, as they are responsible for entering data in BD/RDM, may modify information on already notified in vitro diagnostic medical devices.
The Declarant may be:
Access to the Medical Devices Database/ Repertorio dei Dispositivi Medici has been separated into two paths, depending on whether the registering company has a registered place of business in Italy or not:
Declarants, already enabled to access the Medical Devices Database/ Repertorio dei Dispositivi Medici and already holding the role of "ManufacturerIVD"- “FabbricanteIVD”, will be able to use the same credentials for access, but will have to have the delegation for the registration of In Vitro diagnostic medical devices by each Manufacturer/Authorised Representative.
Operating instructions for the registration of in vitro diagnostic medical devices can be found in the User Manual Medical Device Manufacturer Profile (pdf 9 MB).
If the Manufacturer, or the Authorized representative, or the Delegated person, is an Italian company and intends to carry out the registration directly, it must follow the instructions set out in this section.
Italian companies access to Repertorio dei Dispositivi Medici through the online Medical Devices service, available on Portale delle imprese in the Servizi integrati section of impresa.gov - La mia scrivania.
The legal representatives of the companies concerned are directly authorised to use the service; however, the legal representative of a company may delegate to one or more persons of their choice to operate the online Medical Devices service on their behalf.
To grant the delegation, the legal representative must use the sub-delegation management services available on Portale delle imprese in Servizi integrati section of impresa.gov – Gestione subdeleghe .
At the end of this procedure, functions will be available to the authorised user in order to manage the devices for which he/she is delegated.
The smart card for Italian companies
Access to the online service is allowed to those who are holders of a smart card (such as SPID-Sistema Pubblico di Identità Digitale, CIE-Carta di Identità Elettronica or CNS-Carta Nazionale dei Servizi), the legal representatives and delegates, if any, must be holders of their own smart card.
The smart card is an electronic card designed to hold digital certificates. The digital certificate is a genuine identity document in electronic format which, by certifying the identity of the holder, enables all operations requiring recognition of the holder to be carried out in the "virtual" world. Unlike in the real world, digital certificates are generated by defining their specific purpose in advance: as a result, certificates that can be used to affix electronic signatures are different from certificates that can be used to perform network authentication.
The different types of certificate, also present on a single smart card, are:
The user enabled to use the Medical Devices online service, through Portale delle Imprese must have a smart card containing both the digital authentication certificate and the digital signature certificate.
The use of the smart card requires a specific browser configuration: the relevant technical specifications depend on the type of smart card used and are normally communicated by the smart card provider.
Smart cards can be requested from one of the accredited Certification Authorities, the list of which is available on the DigitPA website in the section Firma digitale - Certificatori accreditati.
Additional ways for Italian companies to register in the BD/RDM that do not provide access via Portale delle imprese are:
Foreign companies can directly register devices in the Medical Device Database/Repertorio dei Dispositivi Medici (BD/RDM) using the online functions, available in both Italian and English.
For registration:
For registration by a foreign company, the following cases arise:
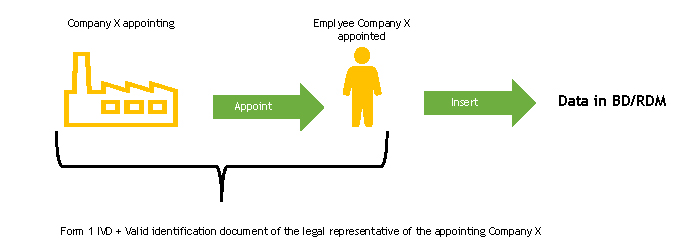
The legal representative of the appointing company must complete the FORM 1-IVD and send it to the Ministry of Health.
The Ministry of Health, upon receipt of the FORM 1-IVD, verifies its completeness.
Following this check, the Ministry of Health registers the person designated to work in the BD/RDM as a user of the NSIS (New Health Information System) Security System and enables him/her to the ManufacturerIVD (FabbricanteIVD) profile.
The person designated to operate the BD/RDM will receive two automatic e-mails at the e-mail address indicated in FORM 1-IVD:
If the person in charge of entering data in the BD/RDM already has access credentials, he/she will only receive the second automatic e-mail, or none at all, if he/she also has the ManufacturerIVD (FabbricanteIVD) role.
At the end of this procedure, functions will be available for the authorised user to enter device data into the BD/RDM. Operating instructions for the registration of Medical Devices can be found in the User Manual Medical Device Manufacturer Profile (pdf, 9 Mb).
In order to use the system, the appointed person must have a qualified electronic signature certificate issued by one of the authorised certifiers in Italy or conforming to the provisions of EU Regulation No. 910/2014- The eIDAS (electronic IDentification Authentication and Signature). The format of the signed data must conform to the PKCS #7 standard.
Foreign companies must use off-line signatures to facilitate the entry of electronically signed files into the system. If problems should arise when performing this operation, the user may contact the servicedesk.salute@smi-cons.it for the necessary checks.
The user will be contacted by e-mail by servicedesk.salute@smi-cons.it for the necessary checks, after which he/she will receive an e-mail informing him/her that he/she can start using the electronic signature in off-line mode to validate the data entered.
Even if the company is not a manufacturer, e.g. an authorised representative, the profile enabling the possibility of entering device data is called "ManufacturerIVD" (“FabbricanteIVD”).
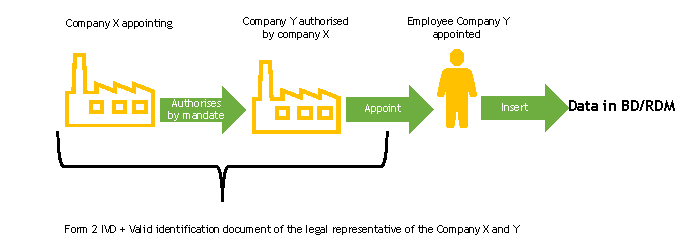
The delegating and declaring company must complete, each for the parts for which it is responsible, the FORM 2-IVD and send it to the Ministry of Health.
The Ministry of Health, upon receipt of the FORM 2-IVD, verifies its completeness.
In the case of a authorised company which is an Italian declarant, it must proceed according to the instructions on the Italian companies page.
In the case of a authorised company which is a foreign declarant, following the check of the form, the Ministry of Health will proceed to register the person designated to work in the BD/RDM as a user of the NSIS Security System and enable him/her to the profile of ManufacturerIVD (FabbricanteIVD).
The person designated to operate the BD/RDM will receive two automatic e-mails at the e-mail address indicated in FORM 2-IVD:
If the person in charge of entering data in the BD/RDM already has access credentials, he/she will only receive the second automatic e-mail, or none at all, if he/she also has the ManufacturerIVD (FabbricanteIVD) role.
The delegating and declaring company enters the duly completed delegation form into the medical device database, following the instructions described in the “Delegation Acquisition” section of the User Manual Medical Device Manufacturer Profile (pdf, 9 Mb).
At the end of this procedure, functions will be available for the authorised user to enter the data of the devices for which he/she has mandate into the database. Operating instructions for the registration of Medical Devices can be found in the User Manual Medical Device Manufacturer Profile (pdf, 9 Mb).
In order to use the system, the appointed person must have a qualified electronic signature certificate issued by one of the authorised certifiers in Italy or conforming to the provisions of EU Regulation No. 910/2014- The eIDAS (electronic IDentification Authentication and Signature). The format of the signed data must conform to the PKCS #7 standard.
Foreign companies must use off-line signatures to facilitate the entry of electronically signed files into the system. If problems should arise when performing this operation, the user may contact the servicedesk.salute@smi-cons.it for the necessary checks.
The user will be contacted by e-mail by servicedesk.salute@smi-cons.it for the necessary checks, after which he/she will receive an e-mail informing him/her that he/she can start using the electronic signature in off-line mode to validate the data entered.
Even if the company is not a manufacturer, e.g. an agent or other delegated company, the profile enabling the possibility of entering device data is called “ManufacturerIVD” (“FabbricanteIVD”).
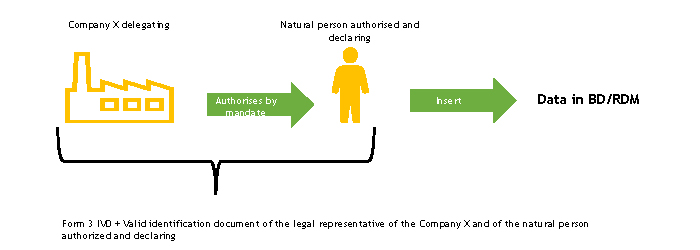
The delegating company and the autorised and declaring natural person must complete, each for the parts for which they are responsible, the FORM 3-IVD and send it to the Ministry of Health.
The Ministry of Health, upon receipt of the FORM 3 -IVD, verifies its completeness.
Following this check, the Ministry of Health registers the person appointed to work in the BD/RDM as a user of the NSIS Security System and enables him/her to the ManufacturerIVD (FabbricanteIVD) profile.
The natural person authorised and declaring to operate in the BD/RDM will receive two automatic e-mails at the e-mail address indicated in FORM 3-IVD:
If the person in charge of entering data in the BD/RDM already has access credentials, he/she will only receive the second automatic e-mail, or none at all, if he/she also has the ManufacturerIVD (FabbricanteIVD) role.
The authorised and declaring natural person enters the duly completed delegation form into the medical device database, following the instructions described in the “Delegation Acquisition” section of the User Manual Medical Device Manufacturer Profile (pdf, 9 Mb).
At the end of this procedure, functions will be available for the authorised user to enter the data of the devices for which he/she has mandate into the database. Operating instructions for the registration of Medical Devices can be found in the User Manual Medical Device Manufacturer Profile (pdf, 9 Mb).
In order to use the system, the designated person must have a qualified electronic signature certificate issued by one of the authorised certifiers in Italy or conforming to the provisions of EU Regulation No. 910/2014- The eIDAS (electronic IDentification Authentication and Signature). The format of the signed data must conform to the PKCS #7 standard.
The natural person must use the off-line signature to facilitate the entry of electronically signed files into the system. If problems should arise when performing this operation, the user may contact the servicedesk.salute@smi-cons.it for the necessary checks.
The user will be contacted by e-mail by servicedesk.salute@smi-cons.it for the necessary checks, after which he/she will receive an e-mail informing him/her that he/she can start using the electronic signature in off-line mode to validate the data entered.
Even if the natural person is not a manufacturer, the profile enabling the possibility of entering device data is called "ManufacturerIVD" (“FabbricanteIVD”).
Whoever makes the notification assumes full responsibility for the information provided, both with regard to general data and individual in vitro diagnostic medical devices.
The publication of the data does not constitute any form of approval by the Ministry of Health.
The General Directorate for Medical Devices and Pharmaceutical Service reserves the right to carry out checks on declarations and registered devices at any time.
All information, initially acquired by the system in processing status "L", must be "validated" through Digital Signature in order to be "published".
The registration activity for each device has a maximum time for completion: once processing has started in the Repertorio, the system allows the "processing" ("L") status to be maintained for a different period of time according to the following cases:
If the system detects the presence in the database of notifications in the processing state, they are listed in the relevant section: Notifications to be completed (To-do List).
The data in the Medical Devices Database/ Repertorio dei Dispositivi Medici (BD/RDM) may only be modified by the person designated as being in charge for entering data in BD/RDM communication, namely the declarant.
User support is divided into several specialised support services for types of problems and clarifications.
At Portale delle imprese, it is possible to receive support and clarification regarding access via the "Medical Devices" online service of the web Portal and regarding the retrieval and use of the smart card.
For further information, send a communication to the certified e-mail address dgdmf@postacert.sanita.it specifying Office 4 - In vitro diagnostic medical devices in the subject line.
For questions concerning the use of the "Company Data" section, you can also contact the Telephone Support Service on 040 3775666 during the following hours:
or to the e-mail address supportoRDM@sanita.fvg.it.
For information on how to access the system via the NSIS platform, on how to use the application and on reporting cases of malfunctioning of IT procedures, contact the Ministry of Health Service Desk:
