Sei in:
Data di pubblicazione: , ultimo aggiornamento
Assistenza sanitaria transfrontaliera: quali diritti per i pazienti
Cross-border healthcare – evaluation of patients’ rights
Siamo inseparabili. Il gatto
4 zampe che cambiano la vita. Una guida all'adozione e alla corretta convivenza con cani e gatti
Per restare sempre insieme. Dimostragli il tuo amore con il microchip
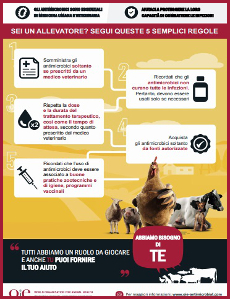
Per un uso consapevole - Antibiotici efficaci
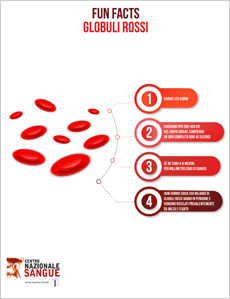
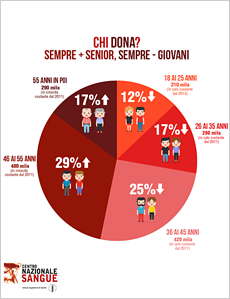
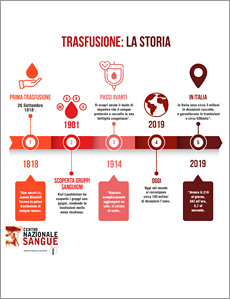
5 buoni motivi per donare il sangue
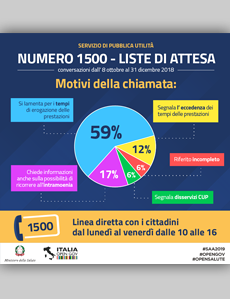
Liste di attesa - Il Piano nazionale 2019-2021 in 10 punti